![]()
![]()
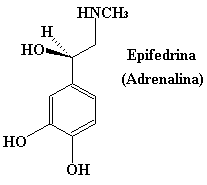
AMINAS FISIOLOGICAMENTE ACTIVAS Un gran número de compuestos químicos con actividad fisiológica contienen nitrógeno. Muchas aminas sencillas se emplean como fármacos. La epifedrina (tambien llamada adrenalina), la propilhexedrina, la hexametilentetramina y la mescalina son algunos ejemplos. Esto por no citar algunas sustancias prohibidas como la muy conocida anfetamina, la metilendioxianfetamina (MDA), mas comunmente conocida como SPEED, y también la metilendioxi-N-metilanfetamina (MDMA) que se denomina en ambitos no académicos como EXTASIS. Una estructura común en muchos de estos compuestos (pero no en todos) es la unidad de 2-fenetilamina. Aunque el mecanismo de estas sustancias no esta aún bien establecido, parece que esa particularidad estructural es necesaria para la unión del compuesto a su receptor.
Estas moléculas no solo son potentes estimulantes del sistema nervioso central, sino que aumentan tambien la actividad cardiovascular y la temperatura corporal, y causan la perdida del apetito. Esta última función es una de la razones de su uso en dietas y tratamiento de la obesidad. Desgraciadamente, pueden producir dependencia química y son potencialmente peligrosas, particularmente cuando se toman sin control médico.
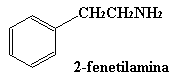
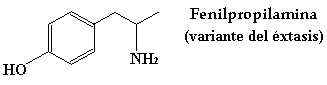
Que no os engańen diciendo que hay muchos tipos de speed (verde, manzana, marrón, rosa,…). El verdadero speed es este:
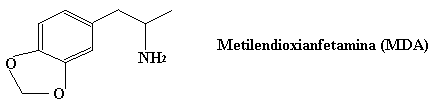
El éxtasis es una variante del speed que se introdujo posteriormente ya que a diferencia del speed no crea adicción, y sólo por la introduccion del grupo metilo (fijaos en la poca diferencia entre ambas estructuras).
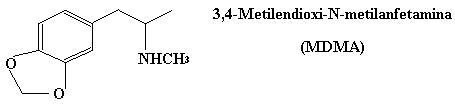
La mescalina es una sustancia alucinógena que se extrae del cactus del peyote que crece en México. Con el peyote también se elabora un licor: el mezcal, aunque su contenido en mescalina es muy bajo. Si veis este licor vereis que contiene dentro de la botella los gusanos que habitan en el cactus. Dicen que quien ingerir los gusanos embriaga más que solamente beber el licor porque los gusanos retienen mayor concentración de mescalina. Realmente, no se que grado de veracidad tiene esta hipótesis.
Hay que tener en cuenta que muchos de estos compuestos tienen distintas funciones fisiológias según cual sea su estereoisomería. Como muchos de vosotros supongo que no estareis familiarizados con este tema os pondre el ejemplo de la anfetamina:
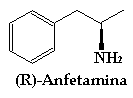
La R-Anfetamina es un potente estimulante del sistema nervioso central, ademas de ser un antidepresivo. La linea gruesa significa que ese enlace está por delante de plano del papel. Sus efectos son muy distintos a los de la S-Anfetamina:
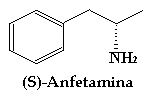
La S-Anfetamina es un estimulante del sistema nervioso simpático, el trazo en discontinua denota que el enlace está por detrás del plano del papel. Solamente me falta comentar que la R-Anfetamina es en la que estais interesados. También hay estereoisómeros de este tipo para el speed y el éxtasis. Si os fijais bien, son muy parecidos y tendran igual significado en este caso:
R significa nitrógeno hacia delante (linea gruesa).
S significa nitrógeno hacia atrás (linea discontinua).
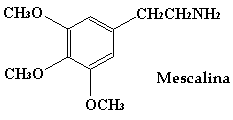
La estereoisomería no es una propiedad que presenten todas las moléculas orgánicas asi por ejemplo, la mescalina que es un potente alucinógeno, no tiene los isómeros R y S. A la hora de sintetizar la anfetamina, es normal obtener una mezcla de ambos isómeros. A estas mezclas se les llama racémicos. Los racémicos deben separarse en sus isómeros.
Muchas aminas, cuyo átomo de nitrógeno forma parte de un anillo (es decir, heterociclos con nitrógeno), también tienen efectos fisiológicos importantes, como por ejemplo la cocaína, la nicotina y la dietilamida del ácido lisérgico (LSD):
La cocaina es un estimulante, anestésico tópico que se encuentra en las hojas de la coca. Genera adicción. Paradójicamente, la dependencia se adquiere antes si en lugar de ser esnifada es fumada.
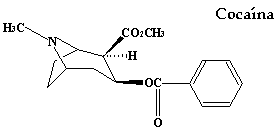
La nicotina se encuentra en una concentración de 2-8% en las hojas secas del tabaco y a pesar de lo que muchos piensan, no es lo que pone los dientes amarillos a los fumadores, el responsable de eso es el alquitrán.
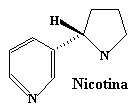
El LSD, sustancia mítica en la década de los sesenta fue descubierto por accidente por el químico suizo Albert Hofmann en los ańos 40 y es la sustancia con mayor poder alucinógeno que se conoce. En su primer autoensayo vivió una experiencia mística totalizadora mientras volvía a su casa montado en bicicleta, de ahi que algunos papeles secantes que se venden en el mercado, impregnados con disoluciones de LSD, muestren una ilustración de una bicicleta.
LA QUIMICA DE LOS ENTEOGENOS INDOLICOS.
Entre las sustancias psicotropas figuran los fármacos o sustancias psicodilésticos, que se caracterizan por su potencia enteógena. El profesor Lehman los divide en cuatro clases:
1. Los derivados catecolamínicos (mescalina, con una estructura básica parecida a la anfetamina y derivados).
2. Los derivados indólicos (LSD, psilocibina, etc).
3. Los anticolinérgicos semejantes al ditrán.
4. Delta-9-tetrahidrocannabinol.
Puesto que el campo de los enteógenos es muy amplio y variado, vamos a referirnos a los derivados indólicos.
La estructura química de gran parte de sustancias enteógenas lleva un núcleo que es un anillo indol. Se trata de una sustancia heterocíclica formada por la unión del benceno y del pirrol. Es, junto con el escatol (beta-metilindol), uno de los productos de la putrefacción de las proteínas y está presente en el excremento del animal. Su nombre de indol se debe a que se encuentra en el colorante índigo. Debido a los dos átomos de N que además llevan en la cadena lateral, la estructura química de estos enteógenos indólicos corresponde a la fundamental de la triptamina.
A propósito de la triptamina, no todos los indoles son enteógenos. Algunos son elementos necesarios en nuestra dieta alimenticia, como el triptofano. Descubierto, como indica su nombre, en la digestión tríptica de los albuminoides, se basa en la estructura heterocíclica del indol. Del triptofano se conocen muchos derivados. Ahora, el más importante es la serotonina o 5-hidroxitriptamina. Como es sabido, la serotonina es un transmisor químico, normal y necesario, de los impulsos nerviosos a través de las sipnasias.
La configuración estructural química de la triptamina puede ser solamente una condición parcial de la acción psicotropa. Para manifestarse como tal, y sobre todo para presentar una acción enteógena, previa de un complemento con sustituciones específicas. Así, mientras la triptamina no desarrolla ningún efecto enteógeno, este empieza a manifestarse en la dietiltriptamina (T-9) y, especialmente, en la dimetiltriptamina (DMT), que llama la atención por su uso en sustitución del LSD, ya que produce una sintomatología enteógena semejante, muy intensa, pero de breve duración.
Jacob ha sintetizado las tres operaciones químicas posible sobre la triptamina y la serotonina:
1. Metilación. Si se metila la función amida terminal de la triptamina, no enteógena, obtenemos la dimetiltriptamina. En cambio si se metila la función amidada terminal de la 5-oxitriptamina o serotonina obtenemos la 5-oxidimetiltriptamina o bufotenina, que se encuentra en el veneno del sapo y es el producto activo del haba de cohoba (Cohoba bello). Tal metilación ejerce efectos enteógenos en el hombre, aparte de que las aminas terciarias pasan mejor la barrera hematoencefálica, actuando directamente sobre el sistema nervioso central.
2. Hidroxilación. En el ciclo bencénico la sustitución por OH en posición 4 origina los productos más enteógenos. A este respecto pueden considerarse la amida lisérgica y lisergamida como una triptamina sustituida en posición 4. El LSD-25 es un producto sintetizado, pero se encuentra en estado natural en las semillas del ololiuqui (los Dondiego). El LSD-25 va perdiendo sus efectos enteógenos a medida que se elimina gradualmente sus grupos etílicos, y, al revés, es interesante comprobar el aumento gradual del enturbamiento de la conciencia. La psilocibina es el éster fosfórico de la psilocina.
3. Oximetilación en 5 sobre la 5-TH: En el cerebro se ha encontrado una hidroxi-indol metil transferasa capaz de fijar in vitro •CH3 sobre el •OH en 5, y volveriamos ha encontrar así la 5-metoxitriptamina (metilserotonina) obtenida en la orina de los reumáticos. Esta sustancia, aunque poco enteógena, es la base de algunos enteógenos.
En los alcaloides de otras plantas mágicas hallaríamos nuevos enteógenos indólicos. La composición química de la ibogaína, alcaloide de la iboga tabernante, volvemos a encontrar el núcleo del indol y, con un poco de buena voluntad, la cadena lateral con 2C y función amida terminal de la triptamina, así como una sustitución •OCH3 en posición 5. Esta misma sustitución, pero ahora en posición 6, son los alcaloides llamados Harmina-yageína y harmalina.
Se incluyen en este grupo de compuestos aquellas sustancias de origen vegetal o animal que contienen en sus moléculas sistemas heterocíclicos nitrogenados y poseen carácter básico. Por esta razón recibieron el nombre de alcaloides; son derivados complejos de la pirina, quinoleina, etc. A pesar de su distinta estructura, poseen propiedades fisiológicas análogas.
Para obtener los alcaloides de los vegetales en que se encuentran, se agotan las partes de la planta que los contienen, reducidas a porciones pequeńas, con agua sola si aquellos se encuentran en forma de sales solubles, o con ácido clorhídrico diluido si están en forma insoluble. Cuando se emplea solamente agua, se trata la disolución obtenida con carbonato sódico; si se emplea como disolvente el ácido clorhídrico, se adiciona a la disolución cal, que pone en libertad el alcaloide. Este se purifica cristalizándolo en el alcohol o éter. Para separar entre sí los diversos alcaloides que pueden coexistir en una disolución se recurre a cristalizaciones fraccionadas y repetidas de sus sales.
La mayor parte de los alcaloides se encuentran en las plantas dicotiledóneas en forma de sales (malatos, tanatos, citratos, etc.). A excepción de algunos como la nicotina, que son líquidos, los restantes son sólidos, poseen sabor amargo y reacción alcalina. Insolubles generalmente en el agua, lo son más en el éter y fácilmente solubles en el alcohol. Con los ácidos forman sales y con ciertos cuerpos dan reacciones, coloreadas o no, pero características. Tienen por lo común, carácter de aminas terciarias, y son en su mayor parte venenos muy violentos; como antídoto se emplea, entre otros, infusiones de té muy cargadas; el tanino de la infusión precipita el alcaloide e impide su asimilación en el tubo digestivo.
Se clasifican los alcaloides teniendo en cuenta los núcleos fundamentales de sus moléculas. Según ellos, se dividen en los grupos siguientes, en los que se hace un estudio somero de los principales alcaloides en ellos contenidos: alcaloides de núcleo pirídico, de núcleo quinoleico, de núcleo isoquinoleico, de núcleo tropánico, de núcleo indólico, de núcleo fenantrénico, y alcaloides de constitución mal definida (según familia de los vegetales, o sea, alcaloides de solanáceas, de papaveráceas, etc... O alcaloides oxigenados o no oxigenados).
A continuación veremos ejemplos de alcaloides dentro de sus diferentes clasificaciones, poniendo como ejemplo los más interesantes o los que más se centran dentro del presente estudio:
1. Alcaloides de núcleo pirídico.
A este grupo pertenecen la nicotina, la pilocarpina, esparteína, entre otros.
Nicotina, C10H15N2. La nicotina se encuentra en el jugo del tabaco acompańada de otros alcaloides. Para su obtención de forma industrial, se hierven las hojas de tabaco con agua varias veces, filtrando y reuniendo los líquidos del tratamiento; se filtran y concentran hasta que el extracto se solidifique y se trata éste con alcohol absoluto y caliente. Por el reposo se forman dos capas, que se separan por decantación; la superior, que contiene la nicotina, se concentra en bańo de María y luego se trata con un exceso de disolución concentrada en potasa, se deja enfriar y se agita con éter, que disuelve la nicotina, la cual se separa del disolvente por evaporación de éste.
La nicotina es un líquido incoloro que hierve a 245 grados, de olor semejante al tabaco y sabor ardiente y picante. Es muy venenosa en dosis extremadas. Sus disolventes son el agua, alcohol, éter, etc...
2. Alcaloides de núcleo isoquinoleico.
Se encuentran estos alcaloides en las plantas papaveráceas y ranunculáceas. El más importante es la papaverina la cual tiene propiedades hipnóticas, aunque no tan acentuadas como la morfina.
3. Alcaloides de núcleo fenatrénico.
El más importante de todos ellos es la morfina de fórmula compleja.
Morfina, C17H19O2N. Se encuentra en el opio en forma de sal, y de él se extrae por varios procedimientos.
La morfina forma cristales rómbicos muy pocos solubles en agua fría, más en la caliente y en el alcohol hirviente. No se emplea pura en Medicina, sino en forma de clorhidrato y sulfato, muy empleados como sedantes y calmantes, dándose en inyecciones hipodérmicas. Deshidratada la morfina, se transforma en apomorfina.
4. Alcaloides de núcleo tropánico.
A este grupo pertenecen, entre otros, dos alcaloides muy importantes: la atropina y la cocaína.
Atropina, C17H22O2N. La atropina es la variedad ópticamente inactiva de la hiosciamina, alcaloide que se encuentra en el jugo de varias plantas como la belladona (Atropa belladona) y el estramonio.
La atropina se obtiene en forma de cristales incoloros poco solubles en agua fría y muy solubles en alcohol, éter, cloroformo, etc. Se emplea muchas veces como sulfato.
Cocaína, C18H21O4N. La cocaína se extrae de las hojas de coca, con el procedimiento que antes explicamos. Se obtiene cristalizada en prismas incoloros de sabor amargo, que insensibilizan la lengua, poco solubles en el agua, y mucho en el alcohol, éter y cloroformo. Se emplea en Medicina en forma de clorhidrato, sal muy soluble en el agua.
5. Alcaloides de núcleo indólico.
Los alcaloides más importantes de este grupo además de los enteógenos, son la estricnina y la brucina.
La forma de obtener los alacaloides indólicos, es siguiendo los pasos que ya detallamos al principio de este capítulo, pues como hemos dicho, los alcaloides son poco solubles en agua, pero muy solubles en alcohol, éter, etc...
Estricnina, C21H22O2N2. Este alcaloide, uno de los más enérgicos, se extrae de diversas plantas, del género Strychnos, entre ellas el haba de San Ignacio, de la nuez vómica, y del jugo de ciertas plantas americanas, pero principalmente de las semillas de la nuez vómica.
Es una sustancia que cristaliza en prismas rómbicos muy poco solubles en agua fría, de sabor amargo muy intenso y muy venenosos, produciendo su ingestión convulsiones tetánicas. La estricnina forma sales con los ácidos, pues tiene marcado carácter alcalino. El sulfato de estricnina es muy usado en terapéutica.
6. Alcaloides de núcleo no definido.
En este grupo se comprenden todos aquellos alcaloides cuya constitución no ha sido aún fijada perfectamente. Entre ellos se encuentra la aconitina, que se encuentra en el acónito (Aconitum napellus). Es un veneno violentísimo, empleado en terapéutica para combatir ciertas dolencias. También encontramos la ergotinina, uno de los principios activos del cornezuelo de centeno, poco soluble en agua, soluble en alcohol y cloroformo, el cual ejerce una acción específica sobre el útero.
Los productos usados para la fabricación de drogas como las anfetaminas, metanfetamina, LSD, metacualona y fenciclidina (PCP), así como de la cocaína y la heroína son, entre otros, la: efedrina, efedrol, fenilpropanona, ergotamina, ácido antralínico, piperidina, éter etílico, amoniaco, alcohol metílico y anhidrido acético.
Como ya hemos visto, la estructura química de la mayoría de los enteógenos, revela un núcleo indol común entre ellos. Este núcleo también aparece en la fórmula de la serotonina, una sustancia que se encuentra en los seres humanos. Es posible que la acción de las drogas enteógenas, psicomiméticas y psicodélicas, se deban a alguna relación con la serotonina en el sistema nervioso central, modificándose o alterándose.
Las fórmulas que a continuación se detallan no son para el «consumo humano», el autor de esta página no se responsabiliza del uso inadecuado o del resultado de las mismas, ya que dichas fórmulas han sido incluidas como mera curiosidad científica, por lo que declina toda responsabilidad.
Si desea hacer algún comentario o tiene fórmulas que desee compartir al resto de los PSICONAUTAS, mándeme un e-mail: b081738103@abonados.cplus.es
- Síntesis del LSD
- Síntesis del LSD (con base ergot)
- Pastillas de litocaina y anfetamina
- Síntesis de MDMA
- Síntesis de la Metanfetamina (speed)
- Kitchen Optimized GHB Synthesis
- 2-CB
Preparatorios preliminares:
El material de partida debe ser algún derivado del ácido lisérgico, del ergot, del grano del centeno, de las semillas de (morning glory), o bien obtenido por alguna vía sintética. Las preparaciones #2 y #3 deben partir de ácido lisérgico solamente, el cual se prepara a partir de su amida como se describe a continuación:
10g de alguna amida de ácido lisérgico obtenida por vía natural se disuelve en 200ml de una disolución de metanol+KOH, y el metanol se elimina inmediatamente al vacio. El residuo es tratado con 200ml de una disolución acuosa de KOH del 8%, y la mezcla es calentada en bańo de vapor durante una hora. Se hace pasar una corriente de nitrógeno en el frasco durante el calentado y el NH3 gas producido se valora con HCl para seguir el proceso. La disolución alcalina debemos neutralizarla con ácido tartárico y como indicador usaremos rojo congo. A continuación la disolución acuosa evaporada se filtra y se limpia por extracción con éter. Se le hace una digestión con metanol para eliminar algunos de los materiales coloreados de los cristales de ácido lisérgico.
Preparamos la luminosidad del laboratorio para que sea similar a una habitación oscura. Debemos tener preparadas luces rojas y amarillas, debido a que los derivados del ácido lisérgico se descomponen en presencia de la luz natural. Debemos usar guantes de goma debido a la naturaleza altamente venenosa de los alcaloides del ergot. Un secador de pelo, o mejor, un evaporador es necesario para acelerar algunos pasos en los que hay que evaporar.
Preparación #1:
Paso 1. Usar luz amarilla.
Colocamos un volumen de alcaloide de ergot pulverizado en un frasco pequeńo y ańadimos dos volumenes de hidracina anhidra. Un procedimiento alternativo es usar un tubo sellado en el que se calientan los reactivos a 112şC. La mezcla se calienta a reflujo durante 30 minutos. Le ańadimos 1.5 volumenes de agua y hervimos 15 minutos. Al introducir la disolución en el frigorifico cristalizará la hidracina del ácido isolisergico.
Paso 2. Usar luz roja.
Enfriamos todos los reactivos y debemos tener hielo cerca para usar. Disolvemos 2.82g de hidracina rapidamente en 100ml de una disolucion 0.1N de HCl que previamente habremos enfriado con hielo. Usaremos un bańo de hielo para mantener el vaso en el que llevamos a cabo la reaccion a 0şC. 100ml de una disolución 0.1N de NaNO2, enfriada con hielo, se ańade a la mezcla y después de 2 o 3 minutos de agitación vigorosa se ańaden 130ml mas de HCl y se agita otra vez en bańo de hielo. Después de 5 minutos de agitación se neutraliza la solución con una disolución saturada de bicarbonato sódico y realizamos una extracción con éter. Eliminamos la disolución acuosa y tratamos de disolver la sustancia viscosa en éter. Ajustamos la disolución de éter por adicción de 3g de dietilamina por cada 300ml de éter extraido. Lo dejamos en ausencia de luz en una habitación que calentamos gradualmente hasta 20şC durante 24 horas. Evaporamos al vacío y tratamos como se indica en la sección de purificación para la conversión de amidas de ácido iso-lisérgico en amidas de ácido lisérgico.
Preparación #2:
Paso 1.Usar luz amarilla.
5.36g de d-ácido lisérgico se suspende en 125ml de acetonitrilo y la suspensión se enfría a unos -20şC en un bańo de acetona enfriada con hielo seco. A la suspensión se le ańade una disolución fría (a -20şC) de 8.82g de anhídrido trifluoroacético disueltos en 75ml de acetonitrilo. Se deja la mezcla a -20şC durante hora y media. En este tiempo los materiales en suspensión se disuelven el d-ácido lisérgico se transforma en la mezcla de anhídrido lisergico y ácido trifluoroacético. El anhídrido puede ser separado en la forma de aceite por evaporación del disolvente al vacío a una temperatura de 0şC, pero esto no es necesario. De todas formas debe permanecer anhídro.
Paso 2. Usar luz amarilla.
La disolución de anhídro mezclado con acetonitrilo del paso 1 se ańade a 150ml de una segunda disolución de acetonitrilo que contiene 7.6g de dietilamina. La mezcla se mantiene en una habitación a temperatura ambiente durante 2 horas. El acetonitrilo se evapora al vacío dejando un residuo de LSD-25 más otras impurezas. El residuo se disuelve en 150ml de cloroformo y 20ml de agua-hielo. El cloroformo se elimina y se le realiza una extracción a la disolución acuosa con varias porciones de cloroformo. Las porciones de cloroformo se combinan para lavarlas con 4 porciones de 50ml de agua-hielo. La disolucion de cloroformo se seca sobre Na2SO4 y se evapora al vacio.
Preparación #3:
Este procedimiento tiene un buen rendimiento y es muy rápido también, origina una pequeńa cantidad de ácido iso-lisérgico (su efecto es medianamente desagradable). De cualquier forma, la estequiometría debe ser exacta o el rendimiento no sera óptimo.
Paso 1. Usar luz blanca.
SO3 es producido en estado anhídro por descomposición de sulfato férrico anhídro a una temperatura de 480şC. Mantenemos SO3 en condiciones anhídras.
Paso 2. Usar luz blanca.
Un recipiente de 22l encajado en un bańo de hielo, condensador, con embudo de llenado y agitador mecánico es llenado con 10 a 11 litros de dimetilformamida (destilada recientemente a presión reducida). El condensador y el embudo de llenado se protegen de la humedad atmosférica. 2 lb de SO3 se introducen con cuidado y se agita muy cautelosamente durante 4 o 5 horas (la adicion de SO3 debe ser poco a poco durante las 4 o 5 horas). La temperatura se mantiene en el intervalo 0-5şC durante la adición. Después de la adición se continua agitando durante 1 o 2 horas hasta que los cristales de sulfuro de trióxido-dimetilformamida se hayan disuelto formando un complejo.
El reactivo se lleva a una pipeta automática cerrada aislada del aire para poner usar la cantidad deseada de reactivos, y los mantenemos frío. Aunque el reactivo, que es coloreado, puede cambiar de amarillo a rojo, lo podemos almacenar en frío durante 3 o 4 meses que permanecerá inalterado. Una licuota de reactivo se disuelve en agua y se valora con NaOH standart hasta el punto final de la fenolftaleína.
Paso 3. Usar luz roja.
Se disuelven de 7.15g de d-ácido lisérgico monohidratado (25mmol) y 1.06g de hidróxido de litio hidratado (25mmol) en 200ml de MeOH preparado. El disolvente es destilado en bańo de vapor a presión reducida. El residuo de lisergato de litio se disuelve en 400ml de dimetilformamida anhidra. De esta disolución se destilan unos 200ml de DMF anhidra a una presión de 15 ml de Hg a través de una columna de hélices de 12 pulgadas. La disolución resultante de lisergato de litio anhidro se enfría a 0şC, con agitación y se trata rápidamente con 500ml de la disolución de SO3-DMF (1.00 molar). La mezcla es agitada en frío durante 10 minutos y luego se ańaden 9.14g (125mmol) de DMF. El agitado y enfriado se prolongan durante 10 minutos más, tras los que se ańaden 400ml de agua. Después se ańaden 200ml de una disolución salina saturada. La amida que se produce es aislada por varias extracciones con porciones de
500ml de dicloroetileno. El combinado que se extrae es secado y luego concentrado hasta que parezca un jarabe a presión reducida. No se debe calentar el jarabe durante la concentración. El LSD cristalizara, pero los cristales y las aguas madres deben ser cromatografiadas de acuerdo con las instrucciones de purificación.
Purificación del LSD:
El material obtenido a partir de cualquiera de las tres preparaciones anteriores puede que contengan ácido lisérgico y amidas del ácido iso-lisérgico. La preparacion #1 contiene en su mayor parte dietilamida del ácido iso-lisérgico que debe ser transformada previa separación. Para esta sustancia, realiza primero el paso 2 de la purificación.
Paso 1. Usar habitacion oscura con luz UV de larga longitud de onda.
El material se disuelve en una mezcla 3:1 de benceno y cloroformo. Carga la columna de cromatografía con una gota de alumina básica en benceno. Una pulgada en la columna equivale a 6 pulgadas de longitud. Saca poco a poco el disolvente hasta el tope de la columna de alumina y ańade cuidadosamente una licuota de disolución de LSD formada por 50ml de disolvente y 1g de LSD. Hazlo correr por la columna seguido por el movimiento más rapido de la banda fluorescente. Después de que este haya sido cogido, limpia el material sobrante de la columna lavandola con MeOH. Usa la luz ultravioleta para prevenir excesivos dańos a los compuestos. Evapora la 2Ş fracción al vacío y ponla a un lado para el paso 2. La fracción que contiene LSD puro se concentra al vacío y el jarabe cristalizara lentamente. Este material se puede convertir en el tartato usando ácido tartárico y el tartato de LSD se cristaliza convenientemente. Su punto de fusión anda por los 190-196şC.
Paso 2. Usar luz roja.
Disuelve el residuo de haber limpiado la columna de cromatografía con metanol en una cantidad minima de alcohol. Ańade dos veces su volumen de una disolución alcohólica con KOH (4N) y deja que la mezcla permanezca a temperatura ambiente durante varias horas. Neutralizala con HCl diluido, hazla ligeramente básica con NH4OH y extrae el producto con cloroformo o dicloroetileno como en las preparaciones #1 y #2. Evapora al vacío y realiza la cromatografía como se indica en el paso 1 de la purificación.
Nota: Los compuestos de ácido lisergico son inestables frente al calor, la luz y el oxígeno. Te puede ayudar a conservarlos ańadir ácido ascórbico como antioxidante, mantener el recipiente bien cerrado, recubierto de laminas de aluminio para evitar la luz y en el frigorifico.
SINTESIS DE LSD (con base de ergot)
Warning!!!!:
Este documento es propiedad de la Cibernetical Implantacion Force, todo lector debe saber ke el presente texto esta escrito solo con fines educativos. Tanto el autor komo la Cibernetical Implantacion Force reniega y declina cualquier responsabilidad civil o penal ke se deriven del uso y/o abuso del presente texto, así que después no me vengas lloriqueando diciéndome en el lío que te he metido,.....esta claro?
Esta síntesis no requiere conocimientos de química ni productos raros, aquí vamos a exponer como hacer LSD desde la cocina de casa, y así os saldrá LSD rico, rico......como diría Arguińano ;-)
El LSD se deriva de los alcaloides del ergot, el cual se encuentra comúnmente en el lúpulo (con el que se hace la cerveza) y en el centeno. La cerveza Foster es una de la pocas que se hacen a partir del centeno y contiene pequeńas cantidades de ergot. No se si esta cerveza esta en el supermercado de la esquina de tu casa, si no esta busca otra que este hecha a partir del centeno y no de la cebada. Ni se te ocurra hacerlo con cerveza hecha a partir de cebada porque después de currartelo no te saldrá LSD.
Pero, ż se podría obtener el ergot de otra manera ? Se podría obtener a través de la morning glory (una planta que no se su nombre en espańol), y de otras semillas que puedes pedir por correo a Hawai. Ahora bien, si lo que esperas es comprarlo en una tienda de productos químicos la verdad es que no se puede. Y si se pudiera no se debe. Un particular puede levantar sospechas comprando productos químicos que sean precursores de drogas. Seré mas claro, te pondré un ejemplo, la metilamina no es precursor de ninguna droga, pero es un compuesto de uso habitual en Sintesis de sustancias psicoactivas, así como en otras Sintesis "legales", bueno pues si te vas a una tienda a comprarla tendrás a la poli vigilándote.
Bueno, compra 12 latas grandes de cerveza Foster (no te las bebas, son la materia prima,... :-D). En una olla calienta la cerveza a fuego lento, esto es para evaporar el agua y concentrar el ergot. Hervir la birra te llevara 36 horas (inventa algún mecanismo o túrnate con tus colegas, al final te sobrara LSD para entripar hasta al gato). Ni se te ocurra acortar el tiempo de ebullición, en tu mano esta obtener LSD o quedarte sin nada. Pero, se podría echar toda la cerveza de golpe y no tener que estar echándola durante 36 horas? La respuesta (y muy a mi pesar) es que no. El hecho de hervir la cerveza durante 36 horas no es solamente evaporarle el agua, sino también otras sustancias menos volátiles que también deben ser eliminadas. Ahora bien, también podrías preguntarte: Si en lugar de 12 latas de cerveza uso 6, żpodría reducir el tiempo a la mitad, es decir a 18 horas? La verdad es que si y no, me explico: Si mantuvieses la ebullición durante 36 horas se podría descomponer tu producto ya que estaría demasiado tiempo en ausencia de agua, pero si lo mantienes 18 horas corres el riesgo de que no se evapore todo lo que hay que eliminar, así que un tiempo prudencial en este caso serian unas 24 horas.
Cuando se te va evaporando la cerveza ańade mas, recuerda que tienes 12 latas. Al final de tanto hervir tienes los componentes esenciales de 12 birras en el fondo de la olla.
Ahora tienes que neutralizar el pH de la disolución de ergotamina que es débilmente ácida, para esto hace falta una base débil. Leche desnatada es ideal para esto. Ańade 1/4 de vaso se leche desnatada (no uses leche normal) y espera 30 minutos a que la reacción alcance el equilibrio. En este punto tienes una disolución de ergotamina con la que puedes seguir el proceso. Esta disolución debe ser de un color marrón cremoso. Pero, al echar la leche żTiene que estar el fuego puesto? No, no tiene que estar al fuego. Como bien he dicho el fuego solo es para concentrar el ergot, no es necesario mantener una temperatura elevada para llevar a cabo una reacción ácido - base. Sin embargo, puede que no ocurra nada contrariante. Si os preguntáis el por qué de usar leche desnatada en lugar de leche normal es porque la leche normal tiene grasas que pueden interaccionar originado reacciones secundarias no deseadas. Ten cuidado porque la ergotamina es un VENENO MORTAL.
Lo siguiente es combinar lisina con la disolución de ergotamina. La lisina la puedes conseguir en cualquier herboristeria. Necesitas alrededor de 2 gramos de lisina (esto es 20 pastillas de 100 mg). Enfría la disolución de ergotamina hasta casi que se congele en tu frigorífico. Ahora muele las pastis de lisina con un mortero, con un molinillo de café o con lo que puedas hasta convertirlas en polvo y ańádeselo a la disolución de ergotamina. Ahora ya tienes ácido lisérgico, que no es poco (muchos que yo se sé darían tortas por el). El ácido lisérgico es ilegal y solo se lo venden a los catedráticos firmando un papel que dice que solo lo usaran para experimentación..........juajuajua >:->
Bueno, tu lo que quieres es LSD que es la dietilamida del ácido lisérgico, así que lo único que tienes que hacer es etilar tu compuesto y luego amoniacarlo. Suena difícil?.....Ahora veras que no!
Simplemente ańade alcohol etílico (del que venden en las farmacias) al ácido lisérgico. Un poco de alcohol puro es lo ideal. Ańade un vaso de alcohol puro y a continuación, para eliminar los hidroxilos ańade 6 vasos de peróxido de hidrogeno al 4% (también le llaman agua oxigenada) y puedes comprarlo en la farmacia que hay al lado de tu casa. Ten cuidadin porque ahora tu compuesto es altamente volátil. Manténlo lejos de fuego o te quemaras las pestańas!
El ultimo paso de la preparación es ańadir 4 onzas de amoniaco puro (28.75gr) a la disolución y déjalo reposar a una temperatura normal durante 3 días. Este tiempo de espera permite que tenga lugar una reacción lentisima. A alta temperatura esta reacción es mas rápida pero te arriesgas a que se pierda el LSD porque se descompone. Al final de los tres días, ya tienes LSD!
Pero, ż todo se hace en la misma olla? żtiene que reposar abierto o cerrado? żen un lugar abierto como un patio puede ser? Todo se hace en una misma olla grande, hay sitio de sobra, pero después al ańadir el agua debes usar un barril (cońo, son 90 litros!). El sitio ideal para dejar reposarlo es una habitación cerrada a oscuras porque el lsd se descompone con la luz, el calor y el oxigeno atmosférico. Se descompone lentamente pero mejor prevenir que curar.......
El rendimiento esperado de LSD es 100 gr, alrededor de un millón de dosis! Debes ser MUY MUY cuidadoso en este punto, porque la disolución que tienes entre manos es extremadamente concentrada. Si te cae en las manos puede que te quedes entripado durante ańos!. Ahora pon 90 litros de agua en un barril vierte el LSD y agítalo con un remo largo. En este nivel de disolución 1/4 de vaso contiene unas 100 dosis. Te puedes servir a tus anchas con un cuentagotas. (3 o 4 gotas por dosis de 50 microgramos, pero eso depende de lo que le guste a cada cual de vosotros). Para evitar tener que usar los 90 litros de un barril, ż se podría hacer en proporción, es decir, si para 100 gr son 90 litros, para 1 gr sería 0,9 litros? Exactamente puedes usar esa ley de las proporciones. Pero el que sean dosis de 50 microgramos no es el indicativo de lo que se debe tomar. El mejor trippi del mercado, el famoso "Panoramix" contiene 200 microgramos. Pero cuando llega al consumidor contiene un poco menos (parte del LSD se habrá descompuesto). Bueno, si tu dices: me comería medio panoramix, pues te tomas 2 dosis de 50 microgramos. De esta forma puedes saber la equivalencia entre lo que has preparado y lo que se vende por ahí.
żQué precauciones se deben tomar para poder obtener LSD de forma casera, es decir, qué fallos son los mas comunes que se podrían cometer?
Casi todos los alcaloides del ergot son altamente venenosos. Cuando el mismo Albert Hoffmann descubrió el LSD-25 no entraba dentro de sus planes probarlo. Lo tomo por accidente y así fue como se desvelaron sus potentes propiedades psicoactivas. Ahora bien, hay que tener sumo cuidado con la Sintesis ya que un pequeńo fallo puede ser mortal. En ella se informa de los pasos mas peligrosos. La ergotamina es un veneno mortal, cuando se etila el ácido lisérgico el compuesto es inflamable, mantenedlo lejos de toda fuente de llama o chispa, y usar un lugar bien ventilado donde no se puedan concentrar los gases (por ejemplo no seria válido un sótano o un garaje). Al diluir la disolución a 90 litros también hay que tener cuidado. Seria muy conveniente usar guantes de goma durante toda la Sintesis, pero especialmente en este punto y cuando se trabaja la ergotamina. Ahh, una última cosa es: żque cońo hago con 1000000 de dosis de LSD? Pues bien, como no creo que tengas la capacidad de consumir y/o distribuir tanta cantidad de LSD (yo al menos no la tengo), el sobrante debéis hervirlo antes de tirarlo por el desagüe. Esto es para que se descomponga el LSD....-Y por que quieres que se descomponga? Cońo, porque el LSD también les afecta a los animales y con tal cantidad se puede causar una verdadera catástrofe medioambiental.
Enjoy It !
A todos mis colegas de la Cibernetical Implantacion Force.
Pastillas de litocaina y anfetamina
Bien kolegas, el otro dia estuve hablando kon un kompańero ke está directamente metido en el mundo de las "pastis"......jejejeje. Bueno, pues este kolega me konto este mejunge ke ahora yo os kuento. No me pregunteis komo ni por ké sobre esto. Yo nunca lo he hecho. Bueno, vayamos al grano:
1ş-Se mezclan 50 mg de benzocaina con 25 mg de dormidina.
La benzocaina es uno de los componentes esenciales, y como bien supondreis no está al alcance de todos (para variar). La puedes conseguir si tienes un amigo que trabaje en un hospital o en una farmacia...jeje
La dormidina es una pastilla para dormir, la venden en las farmacias (aunque no se si necesita receta).
2ş-Por otro lado mezcla 50 mg de litocaina con 50 mg de anfetaminas (rulas).
Ambas cosas las puedes pillar en una farmacia, pero como llegues sin su estricta receta médica te pueden pegar y todo.......jejejeje. Asi que lo mejor es irte a tu camello habitual que tendra estas sustancias...;-)
3ş-Se mezcla todo lo anterior con 5gr de sidrocaina pura.
La sidrocaina si la tienes líquida debes pasarla antes a polvo.żque? żque como lo haces?, pues calentando cońo!!!!! żme dices que donde puedes conseguir sidrocaina?.....pues volvemos al rollo de siempre: es de venta en farmacias si llevas una receta, si no, vete a algun camello que estos tambien trabajan con sidrocaina, creeme.
Total, al final tienes una mezcla en polvo (todos los materiales son sólidos y aqui no ha habido ninguna reaacción química). Esta mezcla vienen a ser unas 20 dosis.
Hasta aqui todo normal, pero despues, este kompańero, que se hace llamar Panoramix (.....jejejeje, bonito nick, me lo deberia haber pillado yo ;-) ) me dijo: Mira Superboom, si vas a hacer esto y quieres mantener una clientela fija, sólo tienes que ańadir 15 mg de matarratas (estricnina) por cada dosis. Con esto conseguiras que tus distinguidos clientes esten totalmente enganchados a tu producto.......
ˇˇżżSerá kabrón el Panoramix este??!!......No solo tiene bastante con cortar las pastis, sino que ademas hace que generen adicción. żque?żme preguntas que que es eso de cortar las pastis?....Pues nada, que la sidrocaina y la litocaina sólo tienen la finalidad de cortar. Algunos camellos (por no decir todos) cogen un gramo de cocaina pura y le ańaden 1/2 gramo de sidrocaina y 1/2 gramo de litocaina. De esta forma consiguen 2 gramos y lo venden como cocaina pura de colombia.....jejeje. Y a esta "tecnica" (por llamarlo de alguna forma, ya que realmente es una putada) le llaman "el corte italiano".
Bueno, para finalizar sólo me falta deciros que este polvo que habeis obtenido, si quereis transformarlo en pastillas sólo teneis que hacer lo siguiente:
Ve a la farmacia y compra el medicamento más barato que vendan sin receta (pero que sean cápsulas). Luego sólo tienes que vaciar las cápsulas, las llenas de tu "producto y depués las cierras.
ˇNo seas mendrugo!........20 dosis--------->20 cápsulas.
*****żque? żque quien ha escrito esto?.....yo no, se escribió solo el otro dia cuando le pegué una patada al ordenador. Se cabreó y el teclado se me avalanzó a la cara. Yo solo me defendí. Fue un acto en defensa propia......jejejeje >;->
La síntesis del éxtasis que exponemos en este texto es una traducción de la que podréis encontrar en el libro pikhal (es la nş 109). Este libro lo podeis encontrar en:
*http://hyperreal.com: Aquí está sólo el tomo 2 que contiene 179 síntesis de derivados anfetamínicos.
*ftp://ursa-major.spdcc.com/pub/pihkal: Aquí están los dos tomos, en el 1ş el autor describe sus experiencias con estas drogas.
A pesar de que este texto es una traducción, está ampliado y mejorado (bueno, eso creo que podreis compararlo vosotros mismos….). La ruta sintética que aquí se expone no es muy usada en los laboratorios clandestinos debido a que se parte del MDA (speed). Sin embargo, puede que el Speed sea el único precursor del éxtasis que podais conseguir. Para mayor información los precursores del éxtasis son:
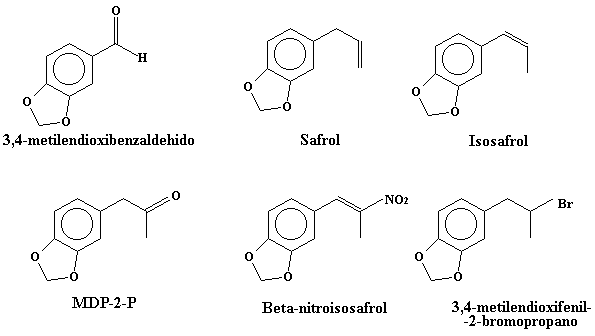
Bueno, vamos al grano:
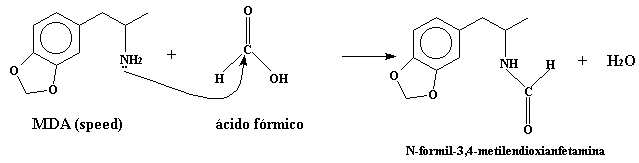
Se disuelven 6.55 gr de MDA (speed) y 2.8 gr de ácido fórmico en 150 ml de benceno, y se lleva a reflujo hasta que no se recoga más agua en el matraz. Unas 20 horas serán suficientes, y se recogeran unos 1.4 ml de agua. Esto es para la formación de la amida. No desespereis en el reflujo, si no estuviese bastante tiempo la disolución a reflujo, se formaria el alcanoato de amonio correspondiente.
Esta es la reaccion que se lleva a cabo en el reflujo:
Para quien no lo sepa, esto es un aparato de reflujo:
Termómetro Columna Claisen
Salida de agua
Refrigerante Liebig
Alargadera
Plato poroso
Entrada de agua
Matraz colector
Matraz de destilación
Manta eléctrica calefactora
A continuación recogemos 8.8 gr de aceite ámbar del que se ha formado, lo lavamos con HCl diluido, luego con NaOH diluido y por último otra vez con HCl diluido. Esto se lleva a cabo en un embudo de decantacion que es un aparato más o menos así:
Al mezclar el aceite ámbar con HCl ó NaOH diluido ocurre como al mezclar agua con aceite: No se mezclan porque sus densidades son diferentes. Por eso los metemos en el embudo de decantación y los agitamos. Luego eliminamos la capa inferior (que es HCl o NaOH, según el caso).
A continuación ańadimos 100 ml de CH2Cl2 y lo eliminamos por extracción al vacío. El resultado esperado son 7.7 gr de N-formil-3,4-metilendioxianfetamina.
Un proceso alternativo para la síntesis de esta amida es mantener a reflujo una disolución de 10 gr de MDA en 20 ml de formato de etilo. Se eliminan las impurezas volátiles (que en este caso es etanol) y se realizan los lavados anteriores. Este proceso alternativo tiene un rendimiento esperado de 7.8 gr de amida.7.7 gr de N-formil-3,4-metilendioxianfetamina pura y seca la disolvemos en 25 ml de tetrahidrofurano (THF) anhidro. Preparamos una disolución de 7.4 gr de hidruro de litio y aluminio (LAH) en 600 ml de THF totalmente anhidro, la agitamos bien y la llevamos a reflujo en atmósfera inerte (por ejemplo con N2). Ahora vertimos la disolucion de N-formil-3,4-metilendioxianfetamina sobre la disolucion de LAH pero con mucho cuidado, y la llevamos a reflujo durante 4 días en atmósfera inerte. El uso de la atmósfera inerte no tiene otra finalidad que la de evitar la humedad ambiental, ya que LAH reacciona violentamente con agua y alcoholes. La reacción es tan exotérmica que es capaz de llevar a cabo la ignición del H2 que se forma. La reacción del LHA con la amida es:
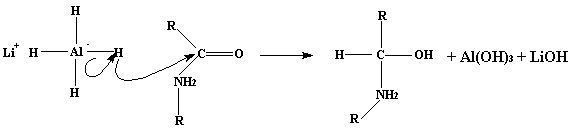
Esto es un mecanismo de reacción general, el rendimiento de este proceso no es muy bueno porque el hidróxido de aluminio es un sólido esponjoso que absorbe parte del compuesto. Asi que, de una reacción de la que cabría esperar el 100% de rendimiento, sólo resulta un 50% de rendimiento. Particularizando esta reaccion a nuestro caso tenemos el siguiente compuesto:
Ahora, antes de enfriar el reflujo hasta temperatura ambiental, el exceso de hidruros se elimina de la disolución ańadiendo una mezcla formada por: 7.4ml de H2O+7.4ml de THF, seguido de 7.4ml de NAOH al 15% de concentración y luego con 22ml de H2O.
El sólido se separa por filtración y se lava el precipitado con THF. Una vez lavado, eliminamos el THF por extracción al vacío y el precipitado lo disolvemos en 200ml de CH2Cl2. A esta disolución se le realiza una extracción con 3 porciones de 100 ml de HCl diluido, y al resultado de la extracción le ańadimos NaOH al 25% de concentración hasta hacer básica nuestra solución. Realizamos otra extracción con 3 porciones de 75 ml de CH2Cl2 para eliminar residuos, y el material no soluble se extrae del disolvente por extracción al vacio. Se habran obtenido unos 6.5 gr de un material blanquecino el cual destilaremos a 100-110şC y a 0.4 mm de Hg de presión, resultando unos 5.0 gr de un aceite incoloro. Lo disolvemos en 25 ml de alcohol isopropílico, lo neutralizamos con ácido clorhídrico concentrado; seguimos con una adición de éter anhidro suficiente para que se vuelva turbia la disolución. En agitación continua, se observará la precipitación de pequeńos cristales de hidrocloruro de 3,4-metilendioxi-N-metilanfetamina (MDMA), que aislaremos por flitración, lavaremos con éter y dejaremos secar, obteniendose finalmente unos 4.8 gr de MDMA.
(de 3,4-metilendioxifeniliacetona) Este es un compuesto intermedio en todos los de la serie MD. Se puede sintetizar a partir de isosafrol o de 3,4-metilendioxibenzaldehido via 1-(3,4-metieldioxifenil)-2-nitropropeno. A una disolución bien agitada de 34 g de peróxido de hidrógeno al 30% en 150 g de ácido fórmico al 80% se le ańade, con cuidado, una disolución de de 32.4 g de isosafrol en 120 ml de acetona. Durante esta adición se produce una reacción exotérmica, y hay que cuidar la cantidad de ácido fórmico vertido porque en ningún momento la temperatura debe rebasar los 40şC. Este paso requiere un poco de tiempo, alrededor de una hora, y un enfriamento externo del vaso con hielo sería muy recomendado. Llevamos la disolución al agitador magnético durante 16 horas, pero teniendo cuidado que la lenta reacción exotérmica no produzca exceso de calor. Para evitar esto, funciona bien un bańo externo en agua corriente. Durante este tiempo, notaremos como vira la disolución de un color naranja a un rojo intenso. Todos los componentes volátiles se eliminan al vacío resultando unos 60 gr de un residuo rojo intenso. Disolvemos el precipitado en 60 ml de metanol, tratamos con 360 ml de H2SO4 al 15% de concentración y lo llevamos a un bańo de vapor durante 36 horas. Después de enfriar, realizamos una extracción con 3 porciones de 75 ml de éter a la mezcla de reacción. El resultado de la extracción se lava primero con agua, y posteriormente con NaOH diluido, y el disolvente se elimina al vacío. El resultado de este proceso lo destilamos a 2mm de Hg de presión a una temperatura de 108-112şC, o a unos 160şC en la bomba de agua. El producto es 20.6 gr de 3,4-metilendioxifenilacetona que es un aceite amarillo pálido. La oxima (de la hidroxilamina) tiene un punto de fusión de 85-88şC y el de la semicarbazona es de unos 162-163şC.
Una síntesis alternativa de la 3,4-metilendioxifenilacetona usa como compuesto precursor al 3,4-metilendioxibezaldehido. Una suspensión de hierro electrolítico en 140 ml de ácido acético glacial se calienta gradualmente en el bańo de vapor. Con bastante calor, pero no mientras haya sales disueltas, se ańade poco a poco, una disolución de 10.0 gr de 1-(3,4-metilendioxifenil)-2-nitropropeno en 75 ml de ácido acético. Controlamos esta adición con una proporción adecuada que permita una reacción vigorosa, pero que no haya demasiada espuma. Observaremos como el color naranja de la mezcla de reacción se torna en un color muy rojizo con la formación de sales blancas y cortezas oscuras. Una vez que hemos completado la adición, seguimos calentando por un periodo de tiempo de una hora y media. Durante este tiempo, la mezcla de reacción se va tornando totalmente blanca y aparecerá un aceite negro en las paredes del vaso. A esta mezcla se le ańaden dos litros de agua y a continuación se extrae con 3 porciones de CH2Cl2 de 100 ml cada una. La parte extraida se lava con varias porciones de NaOH diluido. Después de eliminar el disolvente al vacío, el producto se destila presión reducida a 2mm de Hg de presión a una temperatura de 108-112şC, o a unos 160şC en la bomba de agua. Al final de todo este proceso, habremos obtenido unos 8.0 gr de metilendioxifenilacetona que es un aceite amarillo pálido.
A 40 gr de una lámina delgada de aluminio cortados en cuadrados de una onza (en un erlenmeyer de 2 litro de boca ancha) se le ańaden 1400 ml de agua que contiene 1 gr de cloruro de mercurio. El proceso de la formación de la amalgama será continuo hasta que empiecen a aparecer pequeńas burbujas, la formación de un precipitado gris luminoso y la aparición de puntos plateados en la superficie del aluminio. Este proceso dura entre 15 y 30 minutos dependiendo de lo viejas que sean las superficies del aluminio (conviene usar aluminio recien cortado), de la temperatura del agua y del grosor de la lámina de aluminio (el grosor de las láminas comerciales de aluminio varía de un país a otro). Eliminamos el agua por decantación y el aluminio lo lavamos con dos porciones de 1400 ml de agua fresca. El agua residual del último lavado se elimina totalmente en la medida de lo posible por agitación vigorosa del aluminio. A continuación se ańaden sucesivamente, y mientras agitamos el vaso, 60 gr de hidrocloruro de metilamina disueltos en 60 ml de agua templada, 180 ml de alcohol isopropílico, 145 ml de NaOH al 25% de concentración , 53 gr de 3,4-metilendiofenilxiacetona, y finalmente 350 ml de alcohol isopropílico. Si la forma disponible en que tenemos la metilamina es como base libre en disolución acuosa, la secuencia de adición de los productos será la siguiente: 76 ml de metilamina acuosa al 40%, 180 ml de alcohol isopropílico, una suspensión de 50 gr de NaCl en 140 ml de agua que contiene 25 ml de NaOH 25%, 53 gr de 3,4-metilendiofenilxiacetona, y finalmente 350 ml de alcohol isopropílico. En la mezcla se produce una reacción exotérmica y deberemos mantener la temperatura por debajo de 60şC, para lo cual se hacen necesarios inmersiones ocasionales en agua fria y, cuando se estabilice la temperatura, se deja que la disolución se enfríe hasta llegar a temperatura ambiente. Se observará toda la materia insoluble sedimentada en forma de lodo gris. Decantamos la disolución (que es de un color amarillo) y el lodo lo eliminamos por filtración y lo lavamos con metanol. Al producto decantado, las aguas madres y los lavados les eliminamos el disolvente al vacío. El residuo lo suspendemos con 2400 ml de agua, y suficiente ácido clorhídrico como para que la fase sea ácida. Luego lavamos esto con tres porciones de 75 ml de CH2Cl2, lo hacemos básico con NaOH al 25% y lo extraemos con tres porciones de 100 ml de CH2Cl2. Después de eliminar el disolvente, nos quedaran 55gr de un aceite ámbar que destilaremos a una temperatura de 100-110şC y a una presión de 0.4 mm de Hg produciendo 41 gr de un líquido blanco. Este se disuelve en 200 ml de alcohol isopropílico, se neutraliza con unos 17 ml de HCl concentrado, y luego se trata con 400 ml de éter anhidro. Después filtramos los cristales obtenidos, los lavamos con una mezcla Alcohol isopropílico/éter (2:1), con éter y luego dejamos secar al aire. Habremos obtenido unos 42.0 gr de 3,4-metilendioxi-N-metilanfetamina (MDMA) en forma de finos cristales blancos. La forma de la sal cristalina depende de la temperatura y de la concentración que había en el momento de iniciarse la cristalización. El MDMA obtenido puede ser anhidro o puede ser alguna de sus varias formas hidratadas. Sólo la forma anhidra tiene un punto de fusión a una temperatura definida; los reportajes publicados describen que el punto de fusión de la forma anhidra está en un grado comprendido en el intervalo de 148 a 153şC. Los varios hidratos polimorfos que presenta el MDMA tienen distintos espectros infrarrojos, pero tienen puntos de fusión variables que dependen del grado de calentamiento.
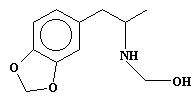
Molécula de MDMA
Dosis: 80-150 mg
Duración: 4-6 horas
SINTESIS DE LA METANFETAMINA (SPEED)
Por Mr.BANDIDO
SINTESIS A PARTIR DE LOS INHALADORES VICKS
Este es el método comprobado y verdadero para extraer metanfetamina a partir de los inhaladores vicks. Estos, contienen "l-desoxiefedrina", lo que es lo mismo que "l-metanfetamina". El l-isómero de la metanfetamina es el relativamente inactivo y se usa como descongestionante suave de la nariz. El d-isómero es que todo el mundo quiere y el que las autoridades han declarado que es demasiado bueno para todo menos para los médicos.
Aquí describo como hacer que funcione el método descrito en la revista "prhack", ya que por si solo unicamente podemos obtener el isómero l (l-met) que es prácticamente inactivo. Para conseguir el d-met, que es el bueno, hay que oxidizar el l-met en fenilacetona, condensarlo con metilamina y finalmente reducirlo.
SINTESIS A PARTIR DE LA ANFETAMINA :
Una de las formas mas sencillas de producir metanfetamina es a partir de la anfetamina. Por supuesto, se asume que en primer lugar tienes anfetamina y que quieres condimentarla un poco.
La diferencia entre la anfetamina y la metanfetamina, es que se ańade un grupo metilo (CH3), al grupo de aminas que se queda suelto en el átomo de carbon, en el centro de la cadena. Por suerte es facil sustituir las aminas. Vaporiza tu amina (anfetamina) con un montón de clorometano (CH3CL, un solvente) vaporizado y un poco de piridina gaseosa. Y ya esta, el grupo amino toma el metilo de clorometano y deja que se vaya el hidrógeno. Este se une al clorino liberado y el HCl resultante se mezcla con la piridina, que es opcional. Si se ańade, se mueve un poco la reacción, sacando el HCl fuera de la ecuación, pero no es necesario.
Si no tienes anfetamina (que es lo mas seguro), otra síntesis posible, es reducir el producto de la condensación de fenilaceto y metilamina. El beneficio de este método es que pueden , aminas diferentes, usarse para producir anfetaminas nobles N-alquil (etanfetamina, butilanfetamina, .....).
3. SINTESIS A PARTIR DE EFEDRINA O PSEUDOEFEDRINA :
Fabricar el speed a partir de la efedrina (que se extrae la planta "efhedra vulgaris", si ves que esto de la quimica de la quimica es muy dificil, hazte una infusión y ya esta ;-)) jejejejeejjeejj, o opseudoefedrina es el método mas utilizado por los laboratorios clandestinos de California. La única diferencia entre metanfetamina (speed) y pseudoefedrina es el grupo alfa-hidroxilo. Si haces reaccionar tu efedrina con el clorido tioxilo, se sustituye el OH con el Cl para producir N-metil.alfa-cloroanfetamina como intermediaria. Es facil hidrogenar este producto : usa hidrido lítico de aluminio, borohidrido sódico o incluso gas hidrógeno con niquel o metal de platino como catalizador. El producto de este paso es N-metilanfetamina y HCl. Deja evaporar el agua y tendras metanfetamina hidroclorido, con lo que ya te puedes ir de fiesta ;-).
SINTESIS A PARTI DE LA FENILAMINA AMINOACIDA :
Es posible una sintesis sencillisima a partir de la fenilamina aminoacida que se puede comprar en cualquier farmacia por unas 1550 pts. La caja lleva 100 tabletas.
La fenilamina es acido 2-amino-3fenilopropanoico, que es mas o menos que una anfetamina con un COOH donde deberia estar el CH3 al final de la cadena. El clorido tionil sustituira al OH con un Cl, que se caera y sera sustituido por H cuando le ańadas hidrido litio aluminio, brohidrido de sodio, gas hidrogeno y niquel/platino. Si usas hidrogeno y un metal para este paso, tendras que reducir el grupo carbonilo con uno de los hidridos. Asi que mejor que ahorres tiempo y esfuerzo y hagas las dos reducciones al mismo tiempo. Cuando se reduce el carbonilo, tienes la anfetamina. Solo tienes que volver al punto numero 2 para convertir la anfetamina en metanfetamina o speed pa la napia .jejejejjeejje.
Espero que con esta informacion a nadie se le ocurra producir grandes cantidades de speed para traficar con lo que aumentarian las asquerosas mafias que en torno él existen, pero si alguien conoce algun metodo facil y casero dentro de lo que se pueda, para producir pequeńas cantidades le agradeceria que lo comunique a la direccion de UNDERHACK en el e-mail de la homepage., y mi colega SUPERBOOM se encargara de lo demas.
Kitchen Optimized GHB Synthesis
Here is a technique which has been optimized for kitchen chemistry.
Ingredients and Materials
gamma-butyrolactone 130ml 98% pure or better NaOH/Red Devil Lye 65gm (plus a couple
extra teaspoons)
distilled water 130ml
activated charcoal 1/4 cup
citric acid 1 or 2 tsp.
substitute: vinegar or HCl (muriatic acid) 1 or 2 tsp.
pH paper (chem. shop, pharmacy, specialty garden shops)
It must be able to measure {very basic, basic, neutral}
glass pyrex dish with lid (min. 2 quart capacity)
substitute: stainless steel (recommend buying the glass)
DONT USE: teflon or aluminum
1 liter bottle (eg coke bottle)
3 plastic or steel spoons (1 to stir, 1 for NaOH, 1 for acid) (the NaOH spoon must be dry)
Optional: distilled water ice cubes
Citric acid can be obtained as "sour salt" in international food stores, stores that cater to Jewish/Arabic cooking, and canning stores.
Hydrochloric acid is sometimes called "muriatic acid" in hardware stores. Low quality HCl can have other gook in it. Either be sure of the quality, or just use vinegar.
NaOH (sodium hydroxide) can be obtained/ordered from photography stores.
Procedure
Mix butyrolactone and NaOH in pyrex dish, and add water. Start breaking up the chunks and mixing the fluids. When it begins to react vigorously, cover it up and let proceed on its own.
Test pH, it should be slightly basic. Add a little NaOH/Lye at a time, stopping when the paper turns very basic. Heat to a simmer to ensure all the butyrolactone is reacted. As it cools, slowly add citric/HCl until the pH is neutral.
Let cool (you can speed this up by carefully adding the ice cubes. Don't let them get on the glass or it might break.) Pour into 1 liter bottle with a quarter cup of activated charcoal. (Wash the charcoal to remove the dust.) Add distilled water to make 1 liter. Let set (shaking now and then) so the charcoal can do its thing.
This works 100%, with no noticable solvant smell in the finished product.
Ingestion
Approx 1 gm GHB per 1 tsp. Start with a low dose and work up, some people get nausea on GHB. About 3 teaspoons for <120 lbs, 4 tsp. <140 lbs, 5 tsp <160 6 tsp < 190 lbs. Increase 1 tsp at a time.
The classic mistake is to take a dose, and, at 15-30 min. you think "wow, this is great but I think I want a little more." More is ingested and you find yourself waking up from having passed out. This can be especially dangerous since the first time you don't know if you will become nauseated. Passed out and puking is a bad, bad combination. (Just ask Jimi Hendrix.)
Always tell your friends if you are on GHB so that if you pass out they will know why and not freak out! This will save you embarassment and big hospital bills.
by,
out-of-it.
4-Bromo-2,5-dimethoxyphenethylamine
A review of this synthesis from a chemist
List of Chemicals
100g 2,5-Dimethoxybenzaldehyde
10g Ammonium acetate
200g Nitromethane
Isopropyl Alchohol (IPA)
500ml Tetrahydrofuran (THF)
Anhydrous Calcium Chloride (eg, Drierite)
NaOH
HCl
20g Bromine
Glacial acetic acid
Methylene chloride
25 grams of LAH
List of Materials
500ml boiling flask
200ml boiling flask
condenser
electric heater/heating mantel
oil bath
boiling chips
filter paper
separation funnel
high-temperature distillation flask
250ml filter flask
2 250ml beakers
500ml beaker
vacuum pump and hose
thermometer
DISCLAIMER #1: It should be perfectly clear that 2-CB, also known under a variety of names such as "venus", "bromo", "erox", or "nexus", is currently a Schedule-I substance. In order to protect us from ourselves, our government has dutifully made it illegal to posses, manufacture, or distribute any amount of 2-CB for any purpose. Any persons caught breaking this law are subject to prosecution, imprisonment, dehumanization, demonization, humiliation, character defamation, intense personal harrasment, and to top it all off - drug diversion. You've been warned.
DISCLAIMER #2: It should be made perfectly clear that laboratory chemistry is a very unforgiving art form. It can take years to master even the simplest of reactions and tiny mistakes can literally blow up in your face. Although this synthesis in not overly complex, it contains some hazards and will take a good deal of laboratory skill, some sophisticated equipment, and a couple of days to complete. Please use caution when testing the final product - it is always best to start off with minute amounts and build dosage slowly so as to minimize any unforeseen adverse effects.
2CB is not exactly easy to make, but it is pretty straightforward. There aren't any very tricky reactions or especially messy procedures and the chemicals are not particularly suspicious to obtain (anymore than anybody buying chemicals or lab equipment today is suspicious to the authorities). But, like any synthesis of this nature, patience and precision are most imperative. Be sure to make the measurements exact, keep temperatures precise and check the colors or consistency of all intermediates. A general chemistry background is also very helpful, but not necessary if you lake the time to learn about what you are doing.
The basic precursor for synthesizing 2CB is 2,5Dimethoxybenzaldebyde (2,5DMB). Although you can even make this compound, the procedure is fairly involved. For our purposes, it is best to start off purchasing this material.
The first procedure is to change the 2,5DMB into 2,5dimethoxynitrostyrene. We do this by heating a mixture of 2,5DMB and nitromethane in the presence of a catalyst, ammonium acetate. Set up an apparatus for refluxing. 'Refluxing' is basically boiling a solvent in such a way that the solvent continually evaporates, condenses, and is returned back to the boiling vessel. To do this we set up the apparatus as shown.
Place 100 Grams of 2,5DMB into a 500ml flask and add 200grams of nitromethane. Most of the solids should dissolve but if they don't immediately, don't worry - they will once you start heating them. When most of them are dissolved, add 10 grams of ammonium acetate. Place the flask into an oil bath on an electric hotplate, connect the condenser and start a slow water flow through the condenser. DO NOT use open flame in any of these procedures. Nitromethane is very flammable! It is, in fact, rocket fuel. It's dangerous enough using an electric hot plate because of the sparks produced when the thermostat clicks on and off, but if you keep reasonable ventilation and a watchful eye on things, you should be okay. Turn the stove up to about 100 degrees celsius, the boiling point of nitromethane, and adjust it until the solution is boiling nicely. In a short time the color of the solution will turn yellow. Four hours later, when you are done refluxing, the color should be a deep red.
Immediately set up for vacuum evaporation. If you let the solution cool off too much it may spontaneously crystalize, making it more difficult to purify, so do a little planning ahead of time. Turn the heat off, detach the condenser from the flask and add a few boiling chips to mixture. Boiling chips provide a very large surface area for lots of small boiling bubbles to form. This will keep the solution boiling smoothly without any "bumps" or big erratic bubbles. Connect the vacuum hose and apply a vacuum very gently at first and build up to a rolling boil without boiling up into the hose. As the solution evaporates, the flask will cool down and you will have to adjust the heat of the hot plate to keep a steady boil going. Eventually, you will remove enough solvent and the whole mass will crystalize into bright pumpkin-orange crystals.
Turn off the heat and remove the vacuum hose from the flask. Always remove the hose from the flask before turning off the water. Otherwise, as the pressure drops water may be sucked into your product. Add 100ml of boiling isopropyl alchohol (IPA) to the mass and stir it as well as you can, breaking up all of the clumps. Your hot plate may still be hot and if you are working near it be sure NOT to splash any of this nitrostyrene onto the hot surface. The resulting fumes are a very potent teargas, something you don't want to experience! Let the flask cool and then pour off the red-colored alcohol solution. Add another 100ml of boiling IPA to the crystals and rinse as before. Rinse at least two more times or until needle-like crystals form out of the amorphous orange mush. Filter these crystals off and air dry completely. There must not be any alchohol left in the crystals, as this will ruin the lithium alumnium hydride (LAH) in the next step. This is 2,5 imethoxynitrostyrene (2,5DMNS).
Dissolve 50 grams of 2,5DMNS in 300ml of anhydrous (dry) THF. If you cannot find anhydrous THF, then you will have to dry it the best you can. Do this by adding a drying agent (such as Drierite, or anhydrous Epson salts) to the THF, let it sit overnight or longer, and then filter it out. This should get most of the moisture out of the THF that would otherwise ruin your expensive LAH.
Arrange your refluxing setup with a 2-liter, 2-neck flask in the oil bath, a magnetic stir bar in the flask and a drying tube attached to the condenser. This can be a spare tube, plugged with cotton and filled with Drierite, that is attached to the open end of the condenser to prevent moisture from the air entering the reaction. Put 750ml of dry THF into the two neck flask and start to stir at a slow speed. SLOWLY add 25 grams of LAH to the stirring THF (LAH is nasty, dangerous stuff. It ignites in the presence of water, it's poisonous to breathe, to touch etc. Read up on it...). If you start adding the LAH too fast it may spark, so take your time. It fuzzes as it hits the THF, so add it carefully and let it easily mix into the THF. Now you are going to add your nitrostyrene solution. This is the reason for the two neck flask. As you add this to the swirling THF, it fizzes up pretty vigorously. Heat is generated and the THF starts to evaporate. Get the water running through the condenser and carefully add a little of the nitrostyrene solution (2,5DMNS) through the extra neck. When the fizzing starts to catch up with you, stopper the neck up quickly to keep the fumes from escaping and let the reaction calm down and cool down a little. Repeat this many times until you have added all of the nitrostyrene. When you are done you should have a dirty gray solution fizzing away nicely. Slowly raise the temperature until reflux is reached and hold it there for 24 hours.
Turn off the heat but leave the stirrer on and let the flask cool down. Once cold, SLOWLY add isopropyl alcohol (IPA) dropwise to neutralize any excess LAH. This will cause considerable fizzing. Make sure you are stirring while you add the IPA; if not, a layer of IPA will form and then any stirring that is done will result in the whole solution jumping right out of the flask. Take your time with this until there is no reaction with the addition of any more IPA.
This gray aluminum slush is nearby impossible to filter out of the solution, but with the addition of 5Oml of 15% NaOH it becomes a white solid, and quite filterable. Now filter out the solids (you will be keeping the THF solution), and then wash the filter cake with a litth extra THF. Combine the THF filtrate and the washings into a boiling flask.
Now you must remove the THF from the mixture. This is done by simple distillation under vacuum. Set up the apparatus as shown. Add a few boiling chips to the flask and place it in the oil bath. Start the vacuum and increase until full strength is reached. As the THF evaporates, the flask cools down and needs more pressure to cause boiling. Finally, use the hot plate to cause steady boiling. Eventually you will be left with a dark-brown/goldenish oily residue. This is very crude 2,5 Dimethoxyphenethylamine, which you need to purify.
Suspend the oily residue in 1 liter of distilled water and make it slightly acidic (pH of around 4) by adding HCl. The 2,5DMPEA will become a water-soluble hydrochloride salt and dissolve into the water; other reaction byproducts will remain water-insoluable and can be easily removed. Add the solution to a suitably sized separation funnel and add 50ml of methylene chloride. Shake vigorously and let the phases separate. Drain out the methylene chloride. Repeat this washing with three more 50ml portions of methylene chloride. This should remove much of the solution's color and it will remain only slightly yellow. We must now freebase the solution before our final purification by fractional distillation.
Make the solution very basic by slowly adding a strong solution of NaOH. The mixture will immediately take on a milky appearance. Shake the mixture vigorously for a few minutes to make sure the 2,5DMPEA HCl is converted to the freebase. This freebase is a dark oil which will separate and start to settle out. Extract the oil by washing, as above, with portions of methylene chloride, but this time keep the washings. Combine all the methylene chloride washings and remove the solvent by distillation as above for removing THF. You will be left with around 50ml of a dark oil.
Now we will distill the oil under reduced pressure and at a relatively high temperature. To do this we use a slightly different setup than the one used to remove the solvents. We won't need the condenser in this case and the flask should be much smaller (around 100 to 250ml) to adequately accommodate the smaller volume of oil. Setup the apparatus as shown. Make sure you use a water trap. Any cold water entering the system will crack the flasks and ruin the procedure. Also make sure to add boiling chips to the flask. Oils tend to really spatter and the chips are absolutely necessary. Bring the system up to full pressure slowly. There will probably still be some methylene chloride that will boil off and end up on the receiving vessel. This is ok. As we heat the system it will eventually evaporate out of the flask. At full water pressure, the oil bath will need to reach 195 Celcius before the 2,5DMPEA will start to come over as clear, waterlike oil. Keep the temperature and pressure constant as the oil slowly drips over. This will take some time depending on your exact setup. When you are satisfied that you have distilled out all the product turn off the heat and disconnect the vacuum from the flask. You should have around 20 grams of pure water-white 2,5DMPEA.
Now we will brominate the 4 position of the benzene ring of our 2,5DMPEA. We do so with pure elemental bromine. Bromine is by far the nastiest stuff you'll ever want to come across. As soon as you open the bottle it will start to crawl out. If you noticed how well packed your bottle of bromine was, you were probably expecting something like this. Use this only in a fume hood if possible. Don't even open the bottle in your house or the whole house will smell like bromine for days and days. Fortunately, we don't have to deal with it for very long. If you have no access to a fume hood (which is probably the case) then at least pick a well venilated area, away from any nosey public, and have a large jar with an airtight seal available. Place your 2,5DMPEA in a 250ml beaker and weigh it on a digital scale. Add to it this same amount of glacial acetic acid. Place another 250ml beaker on the scale, quickly open the bromine and weigh out the same weight as your 2,5DMPEA. Add to it that same amount of glacial acetic acid and quickly add the 2,5DMPEA solution to it. After a few quick stirs, seal it in the large airtight jar. The beaker will heat up but it shouldn't be a problem wrth these quantities. If you scale this up you may need to rig up an external cooling water bath. Light yellow solids will slowly form and in a few hours the solution will cool down. The beaker should pretty much now be solid with light yellow crystals of 2CB-HBr.
This 2CB-HBr has many associated forms involved with it and for a consistent, predictable drug we will need to convert it into the hydrochloride salt. Take the whole mass of 2CB-HBr and filter it. Rinse a few times with cold glacial acetic acid. Place the still-wet crystals into a 500ml beaker and slowly add a 40% NaOH solution. Stir briskly and a dark oil will start to settle out. Separate this oil, as you did above, by extracting with methylene chloride. You will also distill this oil, under pressure and at high temperature, as you did above, and again collect a clear oil. This is pure 2CB freebase.
The final step is making the hydrochloride salt of 2CB. This is done very easily. Since 2CB is not soluble in water, we don't have to worry about completely anhydrous solvents that are usually the case when crystallizing the final products. Add the freebase oil to lOOml of distilled water and slowly add acetic acid while stirring until the freebase dissolves. This won't take very mush acetic acid. Get this solution stirring with the magnetic stirrer and slowly add concentrated HCl. A white precipitate of 2CB-HCl will immediately form. Continue to add the HCl until the solution is thick with fine white crystals. Stop the stirring, and when the crystals settle out, decant the water into another beaker. Add a little water to the crystals, stir and let settle. Add this wash to the beaker with the original decanted solution, put it back on the stirrer and add more HCI. More crystals should form. Repeat this process until you are satisfied that all the crystals have formed. Now add all the crystals together - add water, stir, let settle and decant repeatedly until the wash water has a fairly neutral pH. Now you can filter the crystals and air dry. This is fairly pure 2CB-HCl. Enjoy.
Dosage: in PIHKAL, Shulgin recommends 12-24mg, which is a good range to work with when taken orally. An alternate means of administration is by sniffing. I have found this much more effective. Cut the dosage in half and expect to feel results within 10 minutes or less! Note: Sniffing can be very painful! It's best to do smaller amounts than the whole dose all at once. As know from above, 2CB is not water soluble and will sit in your nose burning for while - and it does burn. But the pain goes away in short time and the results are worth it. Experiment!
------------------------------------------------------------------------
Made possible by: Lycaeum Drug Archives
------------------------------------------------------------------------